В соответствии со статьей 12 Закона Республики Узбекистан «О разрешительных процедурах в сфере предпринимательской деятельности» и постановлением Кабинета Министров Республики Узбекистан от 15 августа 2013 года № 225 «О мерах по реализации Закона Республики Узбекистан «О разрешительных процедурах в сфере предпринимательской деятельности» Кабинет Министров постановляет:
г. Ташкент,
- Настоящее Положение разработано в соответствии с законами Республики Узбекистан «О лекарственных средствах и фармацевтической деятельности» и «О разрешительных процедурах в сфере предпринимательской деятельности» и определяет порядок регистрации и выдачи регистрационного удостоверения лекарственного средства и изделия медицинского назначения.
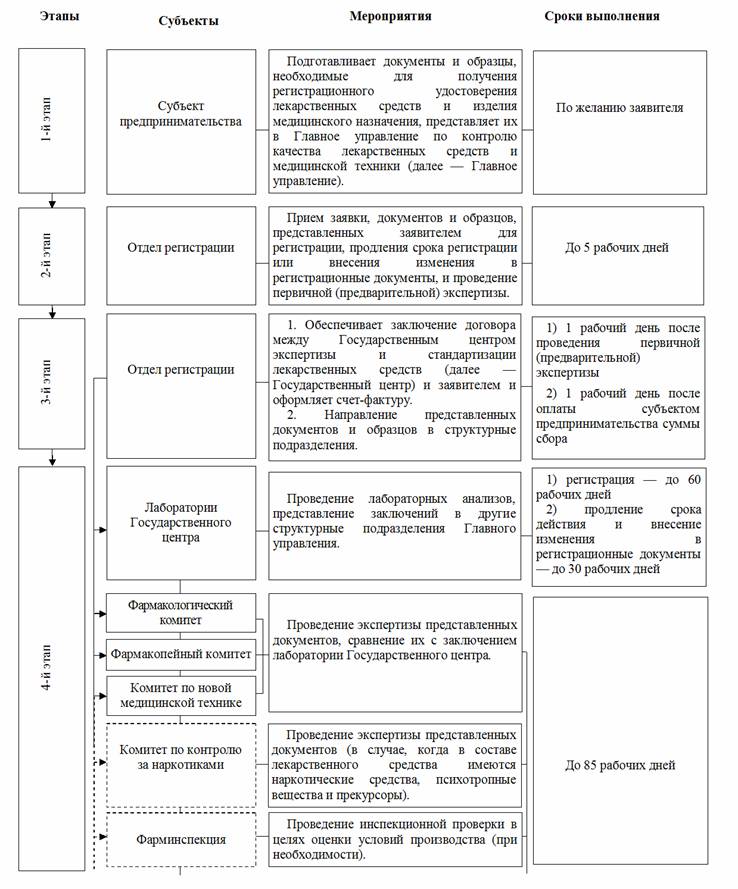
- Регистрация лекарственного средства и изделия медицинского назначения и выдача регистрационного удостоверения осуществляются Главным управлением по контролю качества лекарственных средств и медицинской техники Министерства здравоохранения Республики Узбекистан (далее — Главное управление) по схеме согласно приложению 1 к настоящему Положению.
- Регистрация лекарственного средства и изделия медицинского назначения предусматривает выдачу субъектам предпринимательства, являющимся юридическими лицами (далее — субъекты предпринимательства), регистрационного удостоверения лекарственного средства и изделия медицинского назначения (далее — регистрационное удостоверение).
- Регистрационное удостоверение является основанием для применения лекарственного средства и изделия медицинского назначения в медицинской практике в порядке, установленном законодательными актами.
- Регистрационное удостоверение выдается сроком на 5 лет.
- В случае изменения сведений, приведенных в регистрационных документах лекарственного средства и изделия медицинского назначения, в период действия регистрационного удостоверения на основании представленных субъектом предпринимательства в Главное управление заявления и документов, касающихся изменений, в регистрационные документы лекарственного средства и изделия медицинского назначения вносятся соответствующие изменения и дополнения.
- Регистрация и выдача регистрационного удостоверения на зарубежное лекарственное средство и изделие медицинского назначения осуществляются в соответствии с настоящим Положением.
II. Разрешительные требования и условия
- К разрешительным требованиям и условиям, обязательным к выполнению субъектом предпринимательства при применении в медицинской практике лекарственного средства и изделия медицинского назначения на основании регистрационного удостоверения, относятся:
безусловное соблюдение субъектами предпринимательства, обратившимися за регистрацией лекарственного средства и изделия медицинского назначения, законодательных актов при их применении в медицинской практике;
представление в период действия регистрационного удостоверения в Главное управление владельцем регистрационного удостоверения исчерпывающей информации об изменениях и дополнениях в регистрационные документы, с изложением полных сведений о причине внесения изменений, их влиянии на эффективность, безопасность и показатели качества лекарственного средства и изделия медицинского назначения, включая обязательное регулярное оповещение Главного управления о новых данных касательно фармакологической эффективности и безопасности лекарственного средства;
обязательное соблюдение нормативных документов в области технического регулирования (далее — нормативные документы) на лекарственные средства и изделия медицинского назначения, данных, приведенных в регистрационных документах лекарственных средств и изделий медицинского назначения;
своевременное устранение недостатков, указанных экспертами, а также представление образцов лекарственных средств и изделий медицинского назначения и других материалов в сроки, предусмотренные пунктом 16 настоящего Положения.
III. Документы и образцы, необходимые для получения регистрационного удостоверения
- Для получения регистрационного удостоверения субъект предпринимательства (или доверенное лицо, действующее от его имени) представляет в Главное управление:
а) заявление по форме, согласно приложениям 2 (для лекарственных средств) и 2а (для изделий медицинского назначения) к настоящему Положению;
б) копию свидетельства о государственной регистрации субъекта предпринимательства;
в) регистрационные документы лекарственного средства или изделия медицинского назначения в двух идентичных экземплярах, укомплектованные в порядке, предусмотренном в приложениях 3 (для лекарственных средств) и 3а (для изделий медицинского назначения) к настоящему Положению, сгруппированные по частям, постранично пронумерованные по частям соответственно, заверенные подписью и печатью руководителя субъекта предпринимательства, представляющего заявление;
г) образцы лекарственного средства в количестве, необходимом для проведения трехкратных испытаний в трех промышленных сериях (для зарубежных лекарственных средств одна серия), и изделия медицинского назначения в количестве, необходимом для проведения испытаний в соответствии с нормативными документами, стандартные образцы субстанций (лекарственных веществ), посторонних примесей и родственных веществ, контрольные материалы, специфических реактивов и сертификаты качества на них для определения соответствия лекарственного средства и изделия медицинского назначения требованиям нормативных документов.
- Не допускается требование от субъекта предпринимательства представления документов и образцов, не предусмотренных пунктом 9 настоящего Положения.
- Документы и соответствующие образцы, необходимые для получения регистрационного удостоверения, представляются в Главное управление непосредственно, через средства почтовой связи или в электронной форме с уведомлением об их получении. Документы, представленные в электронной форме, подтверждаются электронной цифровой подписью субъекта предпринимательства.
При этом образцы представляются непосредственно или через средства почтовой связи.
- Документы и соответствующие образцы, представленные в Главное управление для получения регистрационного удостоверения, принимаются по описи, которая незамедлительно выдается (направляется) субъекту предпринимательства с отметкой о дате приема документов, указанием должности, фамилии, имени и подтверждается подписью лица, принявшего документы.
- Конфиденциальные сведения, приведенные в документах, представленных для получения регистрационного удостоверения, не должны разглашаться Главным управлением.
IV. Рассмотрение заявления и принятие решения о выдаче регистрационного удостоверения или об отказе в его выдаче
- Главное управление в установленном порядке через соответствующие отделы осуществляет работу по регистрации лекарственного средства и изделия медицинского назначения.
- За рассмотрение заявления субъекта предпринимательства о выдаче регистрационного удостоверения взимается сбор в 10-кратном размере минимальной заработной платы. Сумма сбора зачисляется на счет Государственного центра экспертизы и стандартизации лекарственных средств (далее — Государственный центр). В случае отказа субъекта предпринимательства от поданного заявления о выдаче регистрационного удостоверения сумма уплаченного сбора возврату не подлежит.
Сумма сбора за рассмотрение заявления зарубежного производителя (или доверенного лица, действующего от его имени) о выдаче регистрационного удостоверения, внесении изменений и дополнений, выдаче дубликата, продлении срока действия, переоформлении регистрационного удостоверения устанавливается Министерством здравоохранения Республики Узбекистан в пределах суммы расходов Главного управления на осуществление указанных процедур.
- Главное управление рассматривает заявление субъекта предпринимательства о выдаче регистрационного удостоверения в срок, не превышающий 180 рабочих дней с даты приема заявления, выдает или отказывает в выдаче регистрационного удостоверения.
В общеустановленный срок регистрации, не превышающий 180 рабочих дней, не включаются:
срок, не превышающий 45 рабочих дней, установленный для устранения недостатков, выявленных в процессе экспертизы лекарственного средства и изделия медицинского назначения, и представления соответствующих документов заявителем;
срок, не превышающий 45 рабочих дней, установленный для представления заявителем в клинические базы программ клинических испытаний, образцов лекарственного средства и изделия медицинского назначения, согласованных в соответствующем порядке для проведения клинических испытаний;
непосредственно сроки проведения клинических испытаний.
- За выдачу регистрационного удостоверения взимается сбор в двукратном размере минимальной заработной платы. Сумма сбора зачисляется на счет Государственного центра.
- Главное управление в соответствии с настоящим Положением по результатам работ по регистрации лекарственного средства и изделия медицинского назначения не позднее 1 рабочего дня с даты принятия соответствующего решения должно выдать субъекту предпринимательства регистрационное удостоверение или уведомить его в письменной форме об отказе в выдаче такого документа.
- Главное управление в целях принятия решения о регистрации лекарственного средства и изделия медицинского назначения в рамках оценки регистрационных документов лекарственного средства и изделия медицинского назначения, условий производства, системы обеспечения качества, соответствия лекарственного средства и изделия медицинского назначения требованиям нормативных документов, качества, эффективности и безопасности, соотношения ожидаемой пользы и риска при применении для здоровья человека может проводить самостоятельно или с привлечением третьих лиц или независимых экспертов нижеследующие экспертные работы, обследования, испытания, анализы, исследования, изучения и научно-техническую оценку в виде:
экспертизы регистрационных документов лекарственного средства и изделия медицинского назначения;
инспекционной проверки с целью оценки и определения соответствия условий производства лекарственного средства и изделий медицинского назначения требованиям правил организации производства и контроля качества, системы управления качеством (в случае внедрения международных стандартов, регулирующих качество лекарственных средств на производственном предприятии);
лабораторных испытаний и анализа лекарственных средств и изделий медицинского назначения;
экспертизы документов лекарственных средств, в составе которых содержатся наркотические средства, психотропные вещества и прекурсоры;
фармакологические, токсикологические исследования;
доклинические исследования, испытание биоэквивалентности.
- Отделы Главного управления при регистрации лекарственного средства или изделия медицинского назначения в порядке и сроки, указанные в приложении 1 к настоящему Положению, осуществляют следующее:
а) отдел регистрации:
после принятия заявления на регистрацию лекарственных средств и изделий медицинского назначения проводит первичную (предварительную) экспертизу (проверку) заявки и прилагаемых к ней образцов и регистрационных документов лекарственных средств и изделий медицинского назначения. В процессе экспертизы рассматривается цель представления заявки и документов, комплект документов в соответствии с лекарственной формой и фармакотерапевтической группой согласно приложениям 3 (для лекарственных средств) и 3а (для изделий медицинского назначения) к настоящему Положению;
ведет соответствующую переписку с заявителем по вопросам регистрации лекарственных средств и изделий медицинского назначения;
на основании положительных результатов первичной (предварительной) экспертизы (проверки) обеспечивает заключение договора между Государственным центром и заявителем и оформляет счет-фактуру;
после оплаты субъектом предпринимательства установленной суммы сбора передает заявление на регистрацию лекарственных средств и изделий медицинского назначения и прилагаемые к нему образцы и регистрационные документы лекарственных средств и изделий медицинского назначения в Государственный центр, Фармакологический, Фармакопейный комитеты, Комитет по новой медицинской технике для проведения экспертизы, а также в Комитет по контролю за наркотиками (в случае, когда в составе лекарственного средства содержатся наркотические средства, психотропные вещества и прекурсоры);
б) лаборатории Государственного центра:
проводят экспертизу административной, химической, фармацевтической, биологической и технической частей регистрационных документов лекарственного средства и изделия медицинского назначения;
оценивают нормативные документы, проводят испытания по определению соответствия образцов лекарственного средства или изделия медицинского назначения требованиям нормативных документов;
передают протоколы испытаний, документы лекарственного средства или изделия медицинского назначения в Фармакопейный, Фармакологический комитеты и Комитет по новой медицинской технике;
в) Фармакопейный комитет:
проводит экспертизу административной, химической, фармацевтической и биологической частей регистрационных документов лекарственного средства, протоколов лабораторных испытаний;
требует от заявителя включения альтернативных методов, дополнительных показателей и методов анализа в нормативные документы или их замены, учитывая современные достижения науки и техники, а также физико-химические свойства действующих и вспомогательных веществ;
проводит экспертизу и повторную экспертизу регистрационных документов с привлечением независимых экспертов;
направляет образцы и документы лекарственного средства в соответствующие лаборатории Государственного центра для дополнительных испытаний в случаях невозможности воспроизведения методов анализа, их недостаточной специфичности, чувствительности и точности, а также внесения изменения в нормативные документы, требующих апробирования методик;
утверждает нормативные документы лекарственных средств и изделий медицинского назначения;
на основании представленных документов и заключений экспертов выносит на Экспертный совет рекомендации о регистрации лекарственных средств или об отказе в их регистрации;
г) Фармакологический комитет:
проводит экспертизу административной, фармакологической, токсикологической и клинической частей регистрационных документов фармакологических и (или) лекарственных средств;
проводит экспертизу и повторную экспертизу документов фармакологических и (или) лекарственных средств с привлечением независимых экспертов;
определяет виды испытаний, одобряет соответствующие клинические базы для проведения клинических испытаний, даёт рекомендации по разработке программы клинических испытаний и одобряет их;
на основании представленных документов и заключений экспертов выносит на Экспертный совет рекомендации о регистрации лекарственных средств без клинических или на основе клинических испытаний или об отказе в их регистрации;
утверждает инструкции по применению лекарственных средств или вносимые в них изменения;
осуществляет деятельность по фармакологическому надзору;
д) Комитет по новой медицинской технике:
проводит экспертизу административной, химической, биологической, технической частей регистрационных документов, протоколов лабораторных анализов, а также экспертизу материалов клинического (медицинского) испытания;
проводит экспертизу и повторную экспертизу регистрационных документов изделий медицинского назначения с привлечением независимых экспертов;
исходя из специфики изделия медицинского назначения, обеспечивает проведение испытаний в аккредитованных лабораториях или на месте установки изделия медицинского назначения;
дает рекомендации по разработке программ клинических (медицинских) испытаний и согласовывает программы клинических (медицинских) испытаний;
определяет виды испытаний, одобряет соответствующие клинические базы для проведения клинических (медицинских) испытаний;
контролирует проведение клинических испытаний изделий медицинского назначения;
утверждает инструкции по применению и маркировку изделий медицинского назначения или вносимые в них изменения или дополнения;
готовит на согласование или утверждение нормативные документы изделий медицинского назначения;
на основании представленных документов и заключений экспертов выносит на Экспертный совет рекомендации о регистрации изделий медицинского назначения без клинических или на основе клинических (медицинских) испытаний или об отказе в их регистрации;
е) Комитет по контролю за наркотиками:
проводит экспертизу регистрационных документов лекарственных средств, имеющих в составе наркотические средства, психотропные вещества и прекурсоры;
при проведении экспертизы лекарственных средств, имеющих в составе наркотические средства, психотропные вещества и прекурсоры, рассматривает наличие данных веществ в списке наркотических средств, психотропных веществ и прекурсоров, находящихся под государственным контролем, разрешения на их медицинское применение, а также необходимость отпуска по рецепту врача и вносит предложения в Фармакологический и Фармакопейный комитеты;
ж) Фарминспекция:
в случае организации производства, контроля качества новой лекарственной формы или вида изделия медицинского назначения проводит обследование условий производства и контроля качества на соответствие правилам организации производства и контроля качества на предприятиях по производству лекарственного средства и изделия медицинского назначения;
по результатам обследования выдает справку о наличии условий производства и контроля качества соответствующего вида лекарственного средства и изделия медицинского назначения;
з) Отдел координации внедрения международных стандартов в фармацевтическую отрасль по результатам инспекции даёт заключение о соответствии предприятия по производству лекарственных средств и изделий медицинского назначения требованиям международных стандартов.
Инспекция осуществляется по решению Экспертного совета, принимаемого на основании заключения экспертов Фармакопейного и Фармакологического комитетов по экспертизе регистрационных документов.
- Экспертный совет на основании заключений Фармакопейного, Фармакологического комитетов, Комитет по новой медицинской технике, а также других подразделений Главного управления в течение семи рабочих дней принимает решение о разрешении или об отказе в применении лекарственного средства и изделия медицинского назначения в медицинской практике.
- Главное управление в течение одного рабочего дня после принятия решения Экспертного совета издает приказ о выдаче регистрационного удостоверения или об отказе в его выдаче.
- Отдел регистрации в соответствии с приказом Главного управления в течение одного рабочего дня оформляет и выдает (или отправляет) субъекту регистрационное удостоверение по формам, согласно приложениям 4 (для лекарственных средств) и 4а (для изделий медицинского назначения) к настоящему Положению.
- Основанием для отказа в выдаче регистрационного удостоверения может быть следующее:
представление субъектом предпринимательства документов, необходимых для выдачи регистрационного удостоверения, образцов лекарственных средств и изделия медицинского назначения и других требуемых материалов, не в полном объеме;
несоответствие субъекта предпринимательства разрешительным требованиям и условиям;
наличие в документах, представленных субъектом предпринимательства, недостоверных или искаженных сведений;
получение обоснованного отрицательного заключения по итогам изучения, исследований, проверок (обследований) или иных научно-технических оценок (экспертизы регистрационных документов лекарственных средств и изделий медицинского назначения, лабораторных или клинических (медицинских) испытаний, характеризующих качество, безопасность и эффективность лекарственных средств и изделий медицинского назначения), осуществление которых в соответствии с настоящим Положением является обязательным.
Отказ в выдаче регистрационного удостоверения по иным основаниям, в том числе по мотивам нецелесообразности, не допускается.
- Уведомление об отказе в выдаче регистрационного удостоверения направляется или выдается (вручается) субъекту предпринимательства в письменной форме с указанием причин отказа, конкретных норм законодательства и срока, в течение которого субъект предпринимательства, устранив указанные причины, может представить документы для повторного рассмотрения. Срок, в течение которого субъект предпринимательства вправе устранить причины отказа и представить документы для повторного рассмотрения, не может быть менее 10 рабочих дней со дня получения уведомления об отказе в выдаче регистрационного удостоверения.
- В случае устранения субъектом предпринимательства причин, послуживших основанием для отказа в выдаче регистрационного удостоверения, в установленный срок повторное рассмотрение документов и продолжение процесса экспертизы осуществляется Главным управлением в срок, не превышающий 10 рабочих дней со дня получения заявления субъекта об устранении причин отказа и соответствующих документов, удостоверяющих устранение причин отказа, за исключением случаев, предусмотренных абзацем третьим настоящего пункта.
За повторное рассмотрение заявления субъекта предпринимательства сбор не взимается.
В случае, если устранение причин отказа субъектом предпринимательства влечет изменение свойств лекарственных средств и изделий медицинского назначения, связанных с их качеством, эффективностью и безопасностью, то заявление считается вновь поданным и рассматривается Главным управлением на общих основаниях.
- При повторном рассмотрении документов не допускается приведение со стороны Главного управления причин отказа, ранее не изложенных в письменной форме заявителю, за исключением приведения причин отказа, связанных с документами, удостоверяющими устранение ранее указанных причин.
- Заявление, поданное субъектом предпринимательства по истечении срока, указанного в уведомлении об отказе в выдаче регистрационного удостоверения, считается вновь поданным и рассматривается Главным управлением на общих основаниях.
- Субъект предпринимательства имеет право обжаловать в установленном порядке отказ в выдаче документа разрешительного характера, а также действия (бездействие) должностного лица Главного управления.
V. Внесение изменений и дополнений в регистрационные документы
- В случае изменения сведений, приведенных в регистрационных документах лекарственного средства и изделия медицинского назначения, в течение срока действия регистрационного удостоверения субъект предпринимательства обращается в Главное управление с заявлением о внесении изменений и дополнений с приложением соответствующих документов, и при необходимости представляет образцы и стандарты.
При этом в случаях возможного негативного действия изменений на качество, эффективность и безопасность лекарственного средства и изделия медицинского назначения Главное управление обоснованно отказывает во внесении изменений и дополнений.
- Заявление субъекта предпринимательства о внесении изменений и дополнений в регистрационные документы в пределах полномочий соответствующих отделов Главного управления рассматривается в срок, не превышающий 90 рабочих дней. За рассмотрение заявления субъекта предпринимательства о внесении изменений и дополнений в регистрационные документы взимается сбор в половинном размере суммы, уплачиваемой за рассмотрение заявления о выдаче регистрационного удостоверения.
- В случае, когда внесение изменения в регистрационные документы требует изменения сведений, приведенных в регистрационном удостоверении, внесение соответствующих изменений в регистрационное удостоверение осуществляется также согласно пунктам 33, 34 и 35 раздела VI настоящего Положения.
VI. Переоформление регистрационного удостоверения, продление срока его действия, выдача дубликатов
- В случае преобразования субъекта предпринимательства, изменения его наименования или местонахождения, без изменения места функционирования (почтового адреса) субъект предпринимательства либо его правопреемник обязан в течение семи рабочих дней после прохождения перерегистрации подать в Главное управление заявление о переоформлении регистрационного удостоверения с приложением документов, подтверждающих указанные сведения.
Документы представляются субъектом предпринимательства в Главное управление непосредственно, либо через средства почтовой связи с уведомлением об их доставке или в электронной форме. Документы, представленные в электронной форме, подтверждаются электронной цифровой подписью субъекта предпринимательства.
Требование от субъекта предпринимательства представления документов, не предусмотренных настоящим пунктом, не допускается.
- До переоформления регистрационного удостоверения субъект предпринимательства или его правопреемник, подавший заявление о переоформлении регистрационного удостоверения, осуществляет применение лекарственного средства или изделия медицинского назначения на основании поданного заявления о переоформлении регистрационного удостоверения с отметкой уполномоченного органа о дате приема заявления.
- При переоформлении регистрационного удостоверения Главное управление вносит соответствующие изменения в реестр выданных регистрационных удостоверений. Переоформление и выдача регистрационного удостоверения осуществляются в срок не более 5 рабочих дней со дня получения Главным управлением заявления о переоформлении регистрационного удостоверения с приложением соответствующих документов.
- За переоформление регистрационного удостоверения взимается сбор в половинном размере суммы, уплачиваемой за рассмотрение Главным управлением заявления субъекта предпринимательства о выдаче регистрационного удостоверения. Сумма сбора зачисляется на счет Государственного центра.
- Срок действия регистрационного удостоверения может быть продлен по заявлению, поданному субъектом предпринимательства в Главное управление с приложением необходимых документов. Заявление о продлении срока действия регистрационного удостоверения должно быть подано в Главное управление в течение трех месяцев до истечения срока его действия. Продление срока действия регистрационного удостоверения осуществляется в порядке, предусмотренном для выдачи регистрационного удостоверения.
За продление срока действия регистрационного удостоверения взимается сбор в половинном размере суммы, уплачиваемой за рассмотрение заявления субъекта предпринимательства о выдаче регистрационного удостоверения. Сумма сбора зачисляется на счет Государственного центра.
- В случае утраты или порчи регистрационного удостоверения по заявлению субъекта предпринимательства выдается его дубликат.
Главное управление обязано выдать или направить дубликат регистрационного удостоверения в срок не более пяти рабочих дней со дня получения заявления, а также оригинала регистрационного удостоверения в случае его порчи и документа, подтверждающего внесение субъектом предпринимательства сбора за выдачу дубликата регистрационного удостоверения.
За выдачу дубликата регистрационного удостоверения взимается сбор в половинном размере суммы, уплачиваемой за рассмотрение Главным управлением заявления субъекта предпринимательства о выдаче регистрационного удостоверения на дату подачи заявления о выдаче дубликата. Сумма сбора зачисляется на счет Государственного центра.
VII. Приостановление, прекращение действия и аннулирование регистрационного удостоверения
- Приостановление, прекращение действия и аннулирование регистрационного удостоверения производится в случаях и порядке, предусмотренных соответственно статьями 22, 23 и 25 Закона Республики Узбекистан «О разрешительных процедурах в сфере предпринимательской деятельности».
- К однократному грубому нарушению разрешительных требований и условий, дающему основание для прекращения в установленном порядке действия регистрационного удостоверения, относятся:
уклонение от проводимых в установленном порядке Главным управлением проверок соблюдения разрешительных требований и условий;
причинение вреда жизни и здоровью граждан либо создание реальной угрозы причинения такого вреда в результате совершения действий и (или) осуществления определенного вида деятельности, на которые выдано регистрационное удостоверение.
VIII. Реестр выданных регистрационных удостоверений
- Главное управление ведет реестр выданных регистрационных удостоверений и размещает его на своих сайтах.
В реестре выданных регистрационных удостоверений указываются следующие сведения о субъектах предпринимательства:
наименование субъекта предпринимательства, его организационно-правовая форма, адрес, телефон;
торговое, международно-непатентованное название лекарственного средства, лекарственная форма, доза, форма выпуска и наименование изделий медицинского назначения, модель, форма, вид, расходные и комплектующие части и др.;
дата выдачи и порядковый номер регистрационного удостоверения;
срок действия регистрационного удостоверения;
основания и даты переоформления, продления, приостановления действия, аннулирования и выдачи дубликата регистрационного удостоверения.
- Информация, содержащаяся в реестре выданных регистрационных удостоверений, является открытой для ознакомления с ней юридических и физических лиц.
к Положению о порядке регистрации лекарственных средств и изделий медицинского назначения и выдачи регистрационного удостоверения
В общеустановленный срок регистрации, не превышающий 180 рабочих дней, не включаются следующие:
срок, не превышающий 45 рабочих дней, установленный для устранения недостатков, выявленных в процессе экспертизы лекарственных средств и изделий медицинского назначения, и представления соответствующих документов заявителем;
срок, не превышающий 45 рабочих дней, установленный для представления образцов лекарственных средств и изделий медицинского назначения, программ клинических испытаний, согласованных в соответствующем порядке, заявителем в клинических базах для проведения клинических испытаний;
непосредственно сроки проведения клинических испытаний.
к Положению о порядке регистрации лекарственных средств и изделий медицинского назначения и выдачи регистрационного удостоверения
(Ф.И.О. руководителя юридического лица Подпись Печать организации)
к Положению о порядке регистрации лекарственных средств и изделий медицинского назначения и выдачи регистрационного удостоверения
(Ф.И.О. руководителя юридического лица Подпись Печать организации)
к Положению о порядке регистрации лекарственных средств и изделий медицинского назначения и выдачи регистрационного удостоверения
*Документы, указанные в перечне, требуются с учетом происхождения, свойств, особенностей способа получения/производства лекарственных средств; если отдельные части документации не включены в регистрационное досье, то следует в соответствующем месте указать причину под соответствующим заголовком.
**При наличии.
***Для лекарственных препаратов животного происхождения в разделе IIС1 должна быть представлена следующая дополнительная информация:
к Положению о порядке регистрации лекарственных средств и изделий медицинского назначения и выдачи регистрационного удостоверения
Выше указанные документы представляются в Главное управление в двух экземплярах.
Копии документов должны быть читабельными и заверены печатью и подписью руководителя организации.
к Положению о порядке регистрации лекарственных средств и изделий медицинского назначения и выдачи регистрационного удостоверения