







Онлайн услуги
-
Опишите подробнее процедуру подписки на журнал
Подписка на журнал осуществляется через АК «Matbuot tarqatuvchi», ОАО “Узбекистон почтаси” и их филиалы, а та...
-
Можно ли скачать журнал в PDF формате?
Да, можно...
Чтобы высказать Ваши предложения, замечания или задать вопрос , пожалуйста, заполните приведенную ниже форму.Подробнее
В данном разделе Вы можете ознакомиться с текущими вакансиями и/или разместить своё резюмеПодробнее
ПОСТАНОВЛЕНИЕ
КАБИНЕТА МИНИСТРОВ РЕСПУБЛИКИ УЗБЕКИСТАН
ОБ УТВЕРЖДЕНИИ ПОЛОЖЕНИЯ О ПОРЯДКЕ РЕГИСТРАЦИИ ЛЕКАРСТВЕННЫХ СРЕДСТВ И ИЗДЕЛИЙ МЕДИЦИНСКОГО НАЗНАЧЕНИЯ И ВЫДАЧИ РЕГИСТРАЦИОННОГО УДОСТОВЕРЕНИЯ
(Собрание законодательства Республики Узбекистан, 2014 г., № 52, ст. 620)
В соответствии со статьей 12 Закона Республики Узбекистан «О разрешительных процедурах в сфере предпринимательской деятельности» и постановлением Кабинета Министров Республики Узбекистан от 15 августа 2013 года № 225 «О мерах по реализации Закона Республики Узбекистан «О разрешительных процедурах в сфере предпринимательской деятельности» Кабинет Министров постановляет:
- Утвердить Положение о порядке регистрации лекарственных средств и изделий медицинского назначения и выдачи регистрационного удостоверения согласно приложению.
- Министерству здравоохранения Республики Узбекистан совместно с заинтересованными министерствами и ведомствами в месячный срок привести ведомственные нормативно-правовые акты в соответствие с настоящим постановлением.
- Контроль за исполнением настоящего постановления возложить на заместителя Премьер-министра Республики Узбекистан А.И. Икрамова.
Премьер-министр Республики Узбекистан Ш. МИРЗИЁЕВ
г. Ташкент,
22 декабря 2014 г.,
№ 352
ПРИЛОЖЕНИЕ
к постановлению Кабинета Министров от 22 декабря 2014 года № 352
ПОЛОЖЕНИЕ
о порядке регистрации лекарственных средств и изделий медицинского назначения и выдачи регистрационного удостоверения
I. Общие положения
- Настоящее Положение разработано в соответствии с законами Республики Узбекистан «О лекарственных средствах и фармацевтической деятельности» и «О разрешительных процедурах в сфере предпринимательской деятельности» и определяет порядок регистрации и выдачи регистрационного удостоверения лекарственного средства и изделия медицинского назначения.
- Регистрация лекарственного средства и изделия медицинского назначения и выдача регистрационного удостоверения осуществляются Главным управлением по контролю качества лекарственных средств и медицинской техники Министерства здравоохранения Республики Узбекистан (далее — Главное управление) по схеме согласно приложению 1 к настоящему Положению.
- Регистрация лекарственного средства и изделия медицинского назначения предусматривает выдачу субъектам предпринимательства, являющимся юридическими лицами (далее — субъекты предпринимательства), регистрационного удостоверения лекарственного средства и изделия медицинского назначения (далее — регистрационное удостоверение).
- Регистрационное удостоверение является основанием для применения лекарственного средства и изделия медицинского назначения в медицинской практике в порядке, установленном законодательными актами.
- Регистрационное удостоверение выдается сроком на 5 лет.
- В случае изменения сведений, приведенных в регистрационных документах лекарственного средства и изделия медицинского назначения, в период действия регистрационного удостоверения на основании представленных субъектом предпринимательства в Главное управление заявления и документов, касающихся изменений, в регистрационные документы лекарственного средства и изделия медицинского назначения вносятся соответствующие изменения и дополнения.
- Регистрация и выдача регистрационного удостоверения на зарубежное лекарственное средство и изделие медицинского назначения осуществляются в соответствии с настоящим Положением.
- К разрешительным требованиям и условиям, обязательным к выполнению субъектом предпринимательства при применении в медицинской практике лекарственного средства и изделия медицинского назначения на основании регистрационного удостоверения, относятся:
- Для получения регистрационного удостоверения субъект предпринимательства (или доверенное лицо, действующее от его имени) представляет в Главное управление:
- Не допускается требование от субъекта предпринимательства представления документов и образцов, не предусмотренных пунктом 9 настоящего Положения.
- Документы и соответствующие образцы, необходимые для получения регистрационного удостоверения, представляются в Главное управление непосредственно, через средства почтовой связи или в электронной форме с уведомлением об их получении. Документы, представленные в электронной форме, подтверждаются электронной цифровой подписью субъекта предпринимательства.
- Документы и соответствующие образцы, представленные в Главное управление для получения регистрационного удостоверения, принимаются по описи, которая незамедлительно выдается (направляется) субъекту предпринимательства с отметкой о дате приема документов, указанием должности, фамилии, имени и подтверждается подписью лица, принявшего документы.
- Конфиденциальные сведения, приведенные в документах, представленных для получения регистрационного удостоверения, не должны разглашаться Главным управлением.
- Главное управление в установленном порядке через соответствующие отделы осуществляет работу по регистрации лекарственного средства и изделия медицинского назначения.
- За рассмотрение заявления субъекта предпринимательства о выдаче регистрационного удостоверения взимается сбор в 10-кратном размере минимальной заработной платы. Сумма сбора зачисляется на счет Государственного центра экспертизы и стандартизации лекарственных средств (далее — Государственный центр). В случае отказа субъекта предпринимательства от поданного заявления о выдаче регистрационного удостоверения сумма уплаченного сбора возврату не подлежит.
- Главное управление рассматривает заявление субъекта предпринимательства о выдаче регистрационного удостоверения в срок, не превышающий 180 рабочих дней с даты приема заявления, выдает или отказывает в выдаче регистрационного удостоверения.
- За выдачу регистрационного удостоверения взимается сбор в двукратном размере минимальной заработной платы. Сумма сбора зачисляется на счет Государственного центра.
- Главное управление в соответствии с настоящим Положением по результатам работ по регистрации лекарственного средства и изделия медицинского назначения не позднее 1 рабочего дня с даты принятия соответствующего решения должно выдать субъекту предпринимательства регистрационное удостоверение или уведомить его в письменной форме об отказе в выдаче такого документа.
- Главное управление в целях принятия решения о регистрации лекарственного средства и изделия медицинского назначения в рамках оценки регистрационных документов лекарственного средства и изделия медицинского назначения, условий производства, системы обеспечения качества, соответствия лекарственного средства и изделия медицинского назначения требованиям нормативных документов, качества, эффективности и безопасности, соотношения ожидаемой пользы и риска при применении для здоровья человека может проводить самостоятельно или с привлечением третьих лиц или независимых экспертов нижеследующие экспертные работы, обследования, испытания, анализы, исследования, изучения и научно-техническую оценку в виде:
- Отделы Главного управления при регистрации лекарственного средства или изделия медицинского назначения в порядке и сроки, указанные в приложении 1 к настоящему Положению, осуществляют следующее:
- Экспертный совет на основании заключений Фармакопейного, Фармакологического комитетов, Комитет по новой медицинской технике, а также других подразделений Главного управления в течение семи рабочих дней принимает решение о разрешении или об отказе в применении лекарственного средства и изделия медицинского назначения в медицинской практике.
- Главное управление в течение одного рабочего дня после принятия решения Экспертного совета издает приказ о выдаче регистрационного удостоверения или об отказе в его выдаче.
- Отдел регистрации в соответствии с приказом Главного управления в течение одного рабочего дня оформляет и выдает (или отправляет) субъекту регистрационное удостоверение по формам, согласно приложениям 4 (для лекарственных средств) и 4а (для изделий медицинского назначения) к настоящему Положению.
- Основанием для отказа в выдаче регистрационного удостоверения может быть следующее:
- Уведомление об отказе в выдаче регистрационного удостоверения направляется или выдается (вручается) субъекту предпринимательства в письменной форме с указанием причин отказа, конкретных норм законодательства и срока, в течение которого субъект предпринимательства, устранив указанные причины, может представить документы для повторного рассмотрения. Срок, в течение которого субъект предпринимательства вправе устранить причины отказа и представить документы для повторного рассмотрения, не может быть менее 10 рабочих дней со дня получения уведомления об отказе в выдаче регистрационного удостоверения.
- В случае устранения субъектом предпринимательства причин, послуживших основанием для отказа в выдаче регистрационного удостоверения, в установленный срок повторное рассмотрение документов и продолжение процесса экспертизы осуществляется Главным управлением в срок, не превышающий 10 рабочих дней со дня получения заявления субъекта об устранении причин отказа и соответствующих документов, удостоверяющих устранение причин отказа, за исключением случаев, предусмотренных абзацем третьим настоящего пункта.
- При повторном рассмотрении документов не допускается приведение со стороны Главного управления причин отказа, ранее не изложенных в письменной форме заявителю, за исключением приведения причин отказа, связанных с документами, удостоверяющими устранение ранее указанных причин.
- Заявление, поданное субъектом предпринимательства по истечении срока, указанного в уведомлении об отказе в выдаче регистрационного удостоверения, считается вновь поданным и рассматривается Главным управлением на общих основаниях.
- Субъект предпринимательства имеет право обжаловать в установленном порядке отказ в выдаче документа разрешительного характера, а также действия (бездействие) должностного лица Главного управления.
- В случае изменения сведений, приведенных в регистрационных документах лекарственного средства и изделия медицинского назначения, в течение срока действия регистрационного удостоверения субъект предпринимательства обращается в Главное управление с заявлением о внесении изменений и дополнений с приложением соответствующих документов, и при необходимости представляет образцы и стандарты.
- Заявление субъекта предпринимательства о внесении изменений и дополнений в регистрационные документы в пределах полномочий соответствующих отделов Главного управления рассматривается в срок, не превышающий 90 рабочих дней. За рассмотрение заявления субъекта предпринимательства о внесении изменений и дополнений в регистрационные документы взимается сбор в половинном размере суммы, уплачиваемой за рассмотрение заявления о выдаче регистрационного удостоверения.
- В случае, когда внесение изменения в регистрационные документы требует изменения сведений, приведенных в регистрационном удостоверении, внесение соответствующих изменений в регистрационное удостоверение осуществляется также согласно пунктам 33, 34 и 35 раздела VI настоящего Положения.
- В случае преобразования субъекта предпринимательства, изменения его наименования или местонахождения, без изменения места функционирования (почтового адреса) субъект предпринимательства либо его правопреемник обязан в течение семи рабочих дней после прохождения перерегистрации подать в Главное управление заявление о переоформлении регистрационного удостоверения с приложением документов, подтверждающих указанные сведения.
- До переоформления регистрационного удостоверения субъект предпринимательства или его правопреемник, подавший заявление о переоформлении регистрационного удостоверения, осуществляет применение лекарственного средства или изделия медицинского назначения на основании поданного заявления о переоформлении регистрационного удостоверения с отметкой уполномоченного органа о дате приема заявления.
- При переоформлении регистрационного удостоверения Главное управление вносит соответствующие изменения в реестр выданных регистрационных удостоверений. Переоформление и выдача регистрационного удостоверения осуществляются в срок не более 5 рабочих дней со дня получения Главным управлением заявления о переоформлении регистрационного удостоверения с приложением соответствующих документов.
- За переоформление регистрационного удостоверения взимается сбор в половинном размере суммы, уплачиваемой за рассмотрение Главным управлением заявления субъекта предпринимательства о выдаче регистрационного удостоверения. Сумма сбора зачисляется на счет Государственного центра.
- Срок действия регистрационного удостоверения может быть продлен по заявлению, поданному субъектом предпринимательства в Главное управление с приложением необходимых документов. Заявление о продлении срока действия регистрационного удостоверения должно быть подано в Главное управление в течение трех месяцев до истечения срока его действия. Продление срока действия регистрационного удостоверения осуществляется в порядке, предусмотренном для выдачи регистрационного удостоверения.
- В случае утраты или порчи регистрационного удостоверения по заявлению субъекта предпринимательства выдается его дубликат.
- Приостановление, прекращение действия и аннулирование регистрационного удостоверения производится в случаях и порядке, предусмотренных соответственно статьями 22, 23 и 25 Закона Республики Узбекистан «О разрешительных процедурах в сфере предпринимательской деятельности».
- К однократному грубому нарушению разрешительных требований и условий, дающему основание для прекращения в установленном порядке действия регистрационного удостоверения, относятся:
- Главное управление ведет реестр выданных регистрационных удостоверений и размещает его на своих сайтах.
- Информация, содержащаяся в реестре выданных регистрационных удостоверений, является открытой для ознакомления с ней юридических и физических лиц.
II. Разрешительные требования и условия
безусловное соблюдение субъектами предпринимательства, обратившимися за регистрацией лекарственного средства и изделия медицинского назначения, законодательных актов при их применении в медицинской практике;
представление в период действия регистрационного удостоверения в Главное управление владельцем регистрационного удостоверения исчерпывающей информации об изменениях и дополнениях в регистрационные документы, с изложением полных сведений о причине внесения изменений, их влиянии на эффективность, безопасность и показатели качества лекарственного средства и изделия медицинского назначения, включая обязательное регулярное оповещение Главного управления о новых данных касательно фармакологической эффективности и безопасности лекарственного средства;
обязательное соблюдение нормативных документов в области технического регулирования (далее — нормативные документы) на лекарственные средства и изделия медицинского назначения, данных, приведенных в регистрационных документах лекарственных средств и изделий медицинского назначения;
своевременное устранение недостатков, указанных экспертами, а также представление образцов лекарственных средств и изделий медицинского назначения и других материалов в сроки, предусмотренные пунктом 16 настоящего Положения.
III. Документы и образцы, необходимые для получения регистрационного удостоверения
а) заявление по форме, согласно приложениям 2 (для лекарственных средств) и 2а (для изделий медицинского назначения) к настоящему Положению;
б) копию свидетельства о государственной регистрации субъекта предпринимательства;
в) регистрационные документы лекарственного средства или изделия медицинского назначения в двух идентичных экземплярах, укомплектованные в порядке, предусмотренном в приложениях 3 (для лекарственных средств) и 3а (для изделий медицинского назначения) к настоящему Положению, сгруппированные по частям, постранично пронумерованные по частям соответственно, заверенные подписью и печатью руководителя субъекта предпринимательства, представляющего заявление;
г) образцы лекарственного средства в количестве, необходимом для проведения трехкратных испытаний в трех промышленных сериях (для зарубежных лекарственных средств одна серия), и изделия медицинского назначения в количестве, необходимом для проведения испытаний в соответствии с нормативными документами, стандартные образцы субстанций (лекарственных веществ), посторонних примесей и родственных веществ, контрольные материалы, специфических реактивов и сертификаты качества на них для определения соответствия лекарственного средства и изделия медицинского назначения требованиям нормативных документов.
При этом образцы представляются непосредственно или через средства почтовой связи.
IV. Рассмотрение заявления и принятие решения о выдаче регистрационного удостоверения или об отказе в его выдаче
Сумма сбора за рассмотрение заявления зарубежного производителя (или доверенного лица, действующего от его имени) о выдаче регистрационного удостоверения, внесении изменений и дополнений, выдаче дубликата, продлении срока действия, переоформлении регистрационного удостоверения устанавливается Министерством здравоохранения Республики Узбекистан в пределах суммы расходов Главного управления на осуществление указанных процедур.
В общеустановленный срок регистрации, не превышающий 180 рабочих дней, не включаются:
срок, не превышающий 45 рабочих дней, установленный для устранения недостатков, выявленных в процессе экспертизы лекарственного средства и изделия медицинского назначения, и представления соответствующих документов заявителем;
срок, не превышающий 45 рабочих дней, установленный для представления заявителем в клинические базы программ клинических испытаний, образцов лекарственного средства и изделия медицинского назначения, согласованных в соответствующем порядке для проведения клинических испытаний;
непосредственно сроки проведения клинических испытаний.
экспертизы регистрационных документов лекарственного средства и изделия медицинского назначения;
инспекционной проверки с целью оценки и определения соответствия условий производства лекарственного средства и изделий медицинского назначения требованиям правил организации производства и контроля качества, системы управления качеством (в случае внедрения международных стандартов, регулирующих качество лекарственных средств на производственном предприятии);
лабораторных испытаний и анализа лекарственных средств и изделий медицинского назначения;
экспертизы документов лекарственных средств, в составе которых содержатся наркотические средства, психотропные вещества и прекурсоры;
фармакологические, токсикологические исследования;
доклинические исследования, испытание биоэквивалентности.
а) отдел регистрации:
после принятия заявления на регистрацию лекарственных средств и изделий медицинского назначения проводит первичную (предварительную) экспертизу (проверку) заявки и прилагаемых к ней образцов и регистрационных документов лекарственных средств и изделий медицинского назначения. В процессе экспертизы рассматривается цель представления заявки и документов, комплект документов в соответствии с лекарственной формой и фармакотерапевтической группой согласно приложениям 3 (для лекарственных средств) и 3а (для изделий медицинского назначения) к настоящему Положению;
ведет соответствующую переписку с заявителем по вопросам регистрации лекарственных средств и изделий медицинского назначения;
на основании положительных результатов первичной (предварительной) экспертизы (проверки) обеспечивает заключение договора между Государственным центром и заявителем и оформляет счет-фактуру;
после оплаты субъектом предпринимательства установленной суммы сбора передает заявление на регистрацию лекарственных средств и изделий медицинского назначения и прилагаемые к нему образцы и регистрационные документы лекарственных средств и изделий медицинского назначения в Государственный центр, Фармакологический, Фармакопейный комитеты, Комитет по новой медицинской технике для проведения экспертизы, а также в Комитет по контролю за наркотиками (в случае, когда в составе лекарственного средства содержатся наркотические средства, психотропные вещества и прекурсоры);
б) лаборатории Государственного центра:
проводят экспертизу административной, химической, фармацевтической, биологической и технической частей регистрационных документов лекарственного средства и изделия медицинского назначения;
оценивают нормативные документы, проводят испытания по определению соответствия образцов лекарственного средства или изделия медицинского назначения требованиям нормативных документов;
передают протоколы испытаний, документы лекарственного средства или изделия медицинского назначения в Фармакопейный, Фармакологический комитеты и Комитет по новой медицинской технике;
в) Фармакопейный комитет:
проводит экспертизу административной, химической, фармацевтической и биологической частей регистрационных документов лекарственного средства, протоколов лабораторных испытаний;
требует от заявителя включения альтернативных методов, дополнительных показателей и методов анализа в нормативные документы или их замены, учитывая современные достижения науки и техники, а также физико-химические свойства действующих и вспомогательных веществ;
проводит экспертизу и повторную экспертизу регистрационных документов с привлечением независимых экспертов;
направляет образцы и документы лекарственного средства в соответствующие лаборатории Государственного центра для дополнительных испытаний в случаях невозможности воспроизведения методов анализа, их недостаточной специфичности, чувствительности и точности, а также внесения изменения в нормативные документы, требующих апробирования методик;
утверждает нормативные документы лекарственных средств и изделий медицинского назначения;
на основании представленных документов и заключений экспертов выносит на Экспертный совет рекомендации о регистрации лекарственных средств или об отказе в их регистрации;
г) Фармакологический комитет:
проводит экспертизу административной, фармакологической, токсикологической и клинической частей регистрационных документов фармакологических и (или) лекарственных средств;
проводит экспертизу и повторную экспертизу документов фармакологических и (или) лекарственных средств с привлечением независимых экспертов;
определяет виды испытаний, одобряет соответствующие клинические базы для проведения клинических испытаний, даёт рекомендации по разработке программы клинических испытаний и одобряет их;
на основании представленных документов и заключений экспертов выносит на Экспертный совет рекомендации о регистрации лекарственных средств без клинических или на основе клинических испытаний или об отказе в их регистрации;
утверждает инструкции по применению лекарственных средств или вносимые в них изменения;
осуществляет деятельность по фармакологическому надзору;
д) Комитет по новой медицинской технике:
проводит экспертизу административной, химической, биологической, технической частей регистрационных документов, протоколов лабораторных анализов, а также экспертизу материалов клинического (медицинского) испытания;
проводит экспертизу и повторную экспертизу регистрационных документов изделий медицинского назначения с привлечением независимых экспертов;
исходя из специфики изделия медицинского назначения, обеспечивает проведение испытаний в аккредитованных лабораториях или на месте установки изделия медицинского назначения;
дает рекомендации по разработке программ клинических (медицинских) испытаний и согласовывает программы клинических (медицинских) испытаний;
определяет виды испытаний, одобряет соответствующие клинические базы для проведения клинических (медицинских) испытаний;
контролирует проведение клинических испытаний изделий медицинского назначения;
утверждает инструкции по применению и маркировку изделий медицинского назначения или вносимые в них изменения или дополнения;
готовит на согласование или утверждение нормативные документы изделий медицинского назначения;
на основании представленных документов и заключений экспертов выносит на Экспертный совет рекомендации о регистрации изделий медицинского назначения без клинических или на основе клинических (медицинских) испытаний или об отказе в их регистрации;
е) Комитет по контролю за наркотиками:
проводит экспертизу регистрационных документов лекарственных средств, имеющих в составе наркотические средства, психотропные вещества и прекурсоры;
при проведении экспертизы лекарственных средств, имеющих в составе наркотические средства, психотропные вещества и прекурсоры, рассматривает наличие данных веществ в списке наркотических средств, психотропных веществ и прекурсоров, находящихся под государственным контролем, разрешения на их медицинское применение, а также необходимость отпуска по рецепту врача и вносит предложения в Фармакологический и Фармакопейный комитеты;
ж) Фарминспекция:
в случае организации производства, контроля качества новой лекарственной формы или вида изделия медицинского назначения проводит обследование условий производства и контроля качества на соответствие правилам организации производства и контроля качества на предприятиях по производству лекарственного средства и изделия медицинского назначения;
по результатам обследования выдает справку о наличии условий производства и контроля качества соответствующего вида лекарственного средства и изделия медицинского назначения;
з) Отдел координации внедрения международных стандартов в фармацевтическую отрасль по результатам инспекции даёт заключение о соответствии предприятия по производству лекарственных средств и изделий медицинского назначения требованиям международных стандартов.
Инспекция осуществляется по решению Экспертного совета, принимаемого на основании заключения экспертов Фармакопейного и Фармакологического комитетов по экспертизе регистрационных документов.
представление субъектом предпринимательства документов, необходимых для выдачи регистрационного удостоверения, образцов лекарственных средств и изделия медицинского назначения и других требуемых материалов, не в полном объеме;
несоответствие субъекта предпринимательства разрешительным требованиям и условиям;
наличие в документах, представленных субъектом предпринимательства, недостоверных или искаженных сведений;
получение обоснованного отрицательного заключения по итогам изучения, исследований, проверок (обследований) или иных научно-технических оценок (экспертизы регистрационных документов лекарственных средств и изделий медицинского назначения, лабораторных или клинических (медицинских) испытаний, характеризующих качество, безопасность и эффективность лекарственных средств и изделий медицинского назначения), осуществление которых в соответствии с настоящим Положением является обязательным.
Отказ в выдаче регистрационного удостоверения по иным основаниям, в том числе по мотивам нецелесообразности, не допускается.
За повторное рассмотрение заявления субъекта предпринимательства сбор не взимается.
В случае, если устранение причин отказа субъектом предпринимательства влечет изменение свойств лекарственных средств и изделий медицинского назначения, связанных с их качеством, эффективностью и безопасностью, то заявление считается вновь поданным и рассматривается Главным управлением на общих основаниях.
V. Внесение изменений и дополнений в регистрационные документы
При этом в случаях возможного негативного действия изменений на качество, эффективность и безопасность лекарственного средства и изделия медицинского назначения Главное управление обоснованно отказывает во внесении изменений и дополнений.
VI. Переоформление регистрационного удостоверения, продление срока его действия, выдача дубликатов
Документы представляются субъектом предпринимательства в Главное управление непосредственно, либо через средства почтовой связи с уведомлением об их доставке или в электронной форме. Документы, представленные в электронной форме, подтверждаются электронной цифровой подписью субъекта предпринимательства.
Требование от субъекта предпринимательства представления документов, не предусмотренных настоящим пунктом, не допускается.
За продление срока действия регистрационного удостоверения взимается сбор в половинном размере суммы, уплачиваемой за рассмотрение заявления субъекта предпринимательства о выдаче регистрационного удостоверения. Сумма сбора зачисляется на счет Государственного центра.
Главное управление обязано выдать или направить дубликат регистрационного удостоверения в срок не более пяти рабочих дней со дня получения заявления, а также оригинала регистрационного удостоверения в случае его порчи и документа, подтверждающего внесение субъектом предпринимательства сбора за выдачу дубликата регистрационного удостоверения.
За выдачу дубликата регистрационного удостоверения взимается сбор в половинном размере суммы, уплачиваемой за рассмотрение Главным управлением заявления субъекта предпринимательства о выдаче регистрационного удостоверения на дату подачи заявления о выдаче дубликата. Сумма сбора зачисляется на счет Государственного центра.
VII. Приостановление, прекращение действия и аннулирование регистрационного удостоверения
уклонение от проводимых в установленном порядке Главным управлением проверок соблюдения разрешительных требований и условий;
причинение вреда жизни и здоровью граждан либо создание реальной угрозы причинения такого вреда в результате совершения действий и (или) осуществления определенного вида деятельности, на которые выдано регистрационное удостоверение.
VIII. Реестр выданных регистрационных удостоверений
В реестре выданных регистрационных удостоверений указываются следующие сведения о субъектах предпринимательства:
наименование субъекта предпринимательства, его организационно-правовая форма, адрес, телефон;
торговое, международно-непатентованное название лекарственного средства, лекарственная форма, доза, форма выпуска и наименование изделий медицинского назначения, модель, форма, вид, расходные и комплектующие части и др.;
дата выдачи и порядковый номер регистрационного удостоверения;
срок действия регистрационного удостоверения;
основания и даты переоформления, продления, приостановления действия, аннулирования и выдачи дубликата регистрационного удостоверения.
ПРИЛОЖЕНИЕ 1
к Положению о порядке регистрации лекарственных средств и изделий медицинского назначения и выдачи регистрационного удостоверения
СХЕМА
регистрации лекарственного средства и изделия медицинского назначения
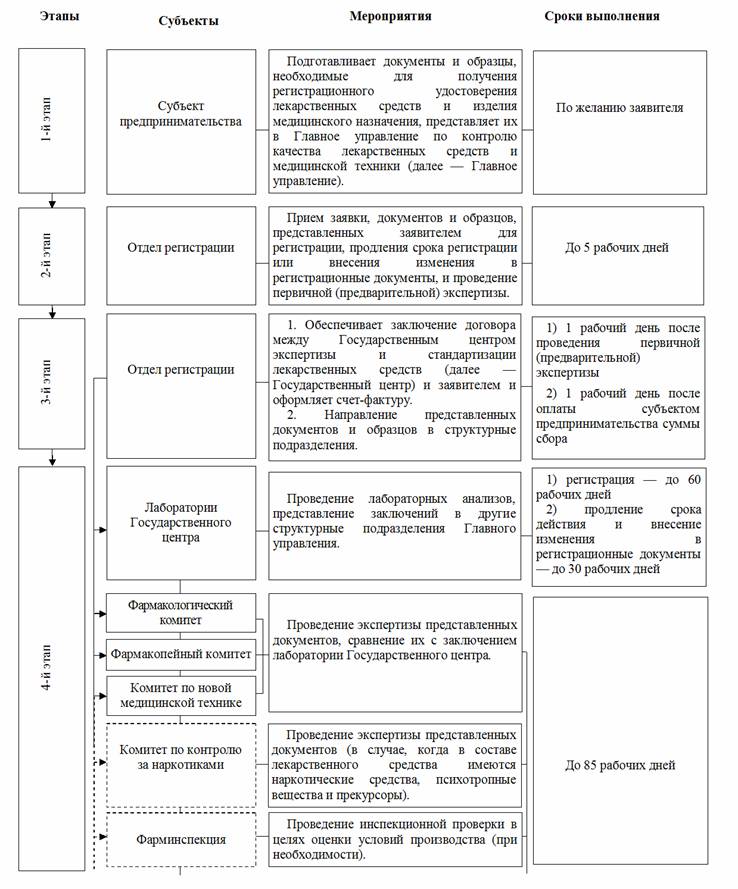

Примечание:
В общеустановленный срок регистрации, не превышающий 180 рабочих дней, не включаются следующие:
срок, не превышающий 45 рабочих дней, установленный для устранения недостатков, выявленных в процессе экспертизы лекарственных средств и изделий медицинского назначения, и представления соответствующих документов заявителем;
срок, не превышающий 45 рабочих дней, установленный для представления образцов лекарственных средств и изделий медицинского назначения, программ клинических испытаний, согласованных в соответствующем порядке, заявителем в клинических базах для проведения клинических испытаний;
непосредственно сроки проведения клинических испытаний.
ПРИЛОЖЕНИЕ 2
к Положению о порядке регистрации лекарственных средств и изделий медицинского назначения и выдачи регистрационного удостоверения
Dori vositasini O‘zbekiston Respublikasida ro‘yxatdan
o‘tkazish uchun
ARIZA
APPLICATION
on registration of a medical product in the Republic
of Uzbekistan
ЗАЯВЛЕНИЕ
на регистрацию лекарственного средства
в Республике Узбекистан
| № | Ma’lumotlar/ Data
(Сведения) | ||
| 1. | Ariza beruvchi tadbirkor to‘g‘risida ma’lumot/Data on the subject of business who is submitting the application
(Сведения о субъекте предпринимательства, подающем заявление) | ||
| Tadbirkorlik subektining nomi, tashkiliy-huquqiy shakli (davlat va ingliz tillarida) / Name, organizational-legal form of the subject of business (in a state and English languages) (Наименование, организационно-правовая форма субъекта предпринимательства (на государственном и английском языках)) | |||
| Pochta adresi va joylashgan manzili (davlat va ingliz tillarida), telefon, faks, е-mail / Mailing address and site (in a state and English languages), phone, fax, e-mail (Почтовый адрес и местонахождение (на государственном и английском языках), телефон, факс, е-mail) | |||
| Rahbar F.I.O./ Full name of the head (Ф.И.О. руководителя) | |||
| 2. | Ishlab chiqaruvchi tashkilot to’g‘risida ma’lumot /Data on a manufacturing organization
(Сведения об организации-производителе) | ||
| Tashkilot nomi, tashkiliy huquqiy shakli (davlat va ingliz tillarida)/ Name, organizational-legal form of the subject of business (in a state and English languages)
(Наименование, организационно-правовая форма субъекта предпринимательства (на государственном и английском языках)) | |||
| Litsenziya raqami, faoliyat turi, amal qilish muddati /Number, period of validity of license, kind of activity
(Номер, срок действия лицензии, вид деятельности) | |||
| Yuridik manzili (davlat va ingliz tillarida),telefon, faks, е-mail/The legal address (in a state and English languages), phone, fax, e-mail
(Юридический адрес (на государственном и английском языках), телефон, факс, е-mail) | |||
| Rahbar F.I.O./Full name of the head
(Ф.И.О. руководителя) | |||
| Ish yuritish bo‘yicha mas’ul shaxs F.I.O. /Full name of the responsible person on office-work
(Ф.И.О. ответственного лица по делопроизводству) | |||
| 3. | Dori vositasi to‘g‘risida ma’lumot/Data on a medical product
(Сведения о лекарственном средстве) | ||
| Dori vositasi nomi (savdo nomi) /Name of a medical product (trade name)
(Наименование лекарственного средства (торговое название)) | Davlat tilida / In a state language (На государственном языке) | ||
| Ingliz tilida / In English language (На английском языке) | |||
| Lotin tilida / In Latin language (На латинском языке) | |||
| Xalqaro patentlanmagan nomi (XPN) /International non proprietary name (INN)
(Международноенепатентованноеназвание (МНН)) | Ingliz tilida / In English language (На английском языке) | ||
| Dori shakli/Medicinal form
(Лекарственная форма) | Davlat tilida/ In a state language (На государственном языке) | ||
| Ingliz tilida/In English language (На английском языке) | |||
| Lotin tilida/In Latin language (На латинском языке) | |||
| Dozasi (mg, ХB, TB va h. k. z.) konsentratsiyasi (mg/ml, XB/ml, ТB/ml va h. k. z.)/
Dose (mg, IU, ЕД etc.) concentration (mg/mL, IU/mL, unit/mLetc.) | |||
| (Доза (мг, МЕ, ЕД и т. д.) коyцентрация (мг/мл, МЕ/мл, ЕД/мл и т. д.)) | |||
| Dori vositasi/Medical product
(Лекарственное средство) |
(Медицинский иммунобиологический препарат (МИБП)) | ||
| 4. | Dori vositasi xom ashyosi to‘g‘risida ma’lumot/Data on raw material of a medical product
(Сведения об исходных продуктах лекарственного средства) | ||
| Xom ashyo turi (keragini belgilang)/Kind of an initial product (mark the necessary)
(Вид исходного продукта(отмечать нужное)) | «in bulk» mahsulot субстанция
«in bulk» product API | ||
| Ta’sir etuvchi modda(lar)ni, ishlab chiqaruvchi tashkilot, davlati/API, manufacturer (s), country(s)
(Действующее (ие) вещество (а), организация-производитель, страна) |
| ||
| Yordamchi moddalar/Additives |
| ||
| 5 | Dori vositasining to‘liq sifat va miqdor tarkibi/Full qualitative and quantitative composition of a medical product
(Полный качественный и количественный состав лекарственного средства) | ||
| Modda/ Substance (Вещество) | Dori shakli birligidagi miqdori/Quantity in one unit of medicinal form (Количествовединицелекарственнойформы) | Sifatini belgilovchi normativ hujjat /Normative document regulating the quality (Нормативныйдокумент, регламентирующийкачество) | |
| Ta’sir etuvchi modda(lar)/Active ingredient(s)
(Действующее(ие) вещество(а)) | |||
| 1. Ta’sir etuvchi modda nomi/ Name of API | |||
| 2. | |||
| 3. | |||
| Yordamchi modda (lar)/Additives
(Вспомогательное(ые) вещество (а)) | |||
| 1.Yordamchi modda nomi/name of an additive | |||
| 2. | |||
| 3. | |||
| 6. | Dorivor o‘simlik xomashyosi (yig‘ma)/Combination herbal medicinal product
(Лекарственное растительное сырье (сбор)) | ||
| O‘simliklarning botanik lotincha nomi, o‘zbekcha va inglizcha nomi (yig‘ma tarkibiga кiruvchi)
/Botanical Latin names of plants, names in Uzbek and English languages (combination herbal medicinal product) (Ботанические латинские названия растений, названия на узбекском и английском языках (входящих в состав сбора)) | Sifatini reglamentlovchi me’yoriy hujjat/Normative document regulating the quality
(Нормативный документ, регламентирующий качество) | Ishlab chiqaruvchi korxona nomi va manzili/The name and address of manufacturer
(Наименование и адрес организации-производителя) | |
| 7. | Dori vositasi o‘rami to‘g‘risida ma’lumot (mavjudligi va qisqa tavsifi)/Data on packing of a medical product
(Сведения об упаковке лекарственного средства) | |
| Birlamchi (dori vositasi miqdori ko‘rsatilgan holda)/Innerpack (with note of quantity of a medical product)
(Первичная (с указанием количества лекарственного средства)) | ||
| Ikkilamchi (birlamchi o‘ramlar ko‘rsatilgan holda)/ Outerpack(with note of quantity of inner packs)
(Вторичная (с указанием количества первичных упаковок)) | ||
| Guruhli (ikkilamchi o‘ramlar ko‘rsatilgan holda)/ Grouppack(with note of quantity of outer packs)
(Групповая (с указанием количества вторичных упаковок)) | ||
| 8. | Farmakologik ma’lumotlar/Pharmacological information
(Фармакологические сведения) | |
| Asosiy farmakologik ta’siri/Main pharmacological action
(Основное фармакологическое действие) | ||
| Farmakoterapevtik guruhi/Pharmacotherapeutic group
(Фармакотерапевтическая группа) | ||
| Qo‘llash sohasi (dori vositasining tibbiyot amaliyotida qo‘llash tavsiya etilgan kasalliklar ko‘rsatilgan holda/A scope (specify the diseases at which a medical product is recommended)
(Область применения (с указанием заболеваний, при которых рекомендовано медицинское применение лекарственного средства)) | ||
| АТХ kodi (mavjud bo‘lganda) yoki unga tegishli takliflar/ ATCa code or if necessary offers concerning it
(Код АТХ (при наличии) или предложения касательно его) | ||
| 9. | Dori vositasining yaroqlilik muddati to‘g‘risida ma’lumot/Data on shelf-life of a medical product
(Сведения о сроке годности лекарственного средства) | |
| Yaroqlilik muddati/Shelf-life
(Срок годности) | ||
| Birlamchi o‘ramni birinchi marotaba ochilgandan yoki eritilgandan so‘ng saqlash (yaroqlilik muddati) /Shelf-life (use) after the first opening of inner packing or dissolution of contents of inner packing
(Срок годности (использования) после первого вскрытия первичной упаковки или растворения содержимого первичной упаковки) | ||
| 10. | Dori vositasini saqlash to‘g‘risida ma’lumot/Storage conditions of a medical product
(Сведения об условиях хранения лекарственного средства) | |
| 11. | Dori vositasini dorixonadan berish tartibi/ Form of delivery of a medical product
(Условия отпуска лекарственного средства из аптеки) | |
To‘ldirilgan sana:
20 yil «_____»_________
Yuridik shaxs rahbarining F.I.O.
Imzo
Tashkilot muhri
Date of filling: (Дата заполнения):
«_____»_________ 20 г
Full name of the Нead
Signature
Organization stamp
(Ф.И.О. руководителя юридического лица Подпись Печать организации)
ПРИЛОЖЕНИЕ 2а
к Положению о порядке регистрации лекарственных средств и изделий медицинского назначения и выдачи регистрационного удостоверения
Tibbiy buyumlarni O‘zbekiston Respublikasida
ro‘yxatdan o‘tkazish uchun
ARIZA
APPLICATION
on registration of a medical device in the Republic
of Uzbekistan
ЗАЯВЛЕНИЕ
на регистрацию изделия медицинского назначения
в Республике Узбекистан
| № | Ma’lumotlar/Data
(Сведения) | |
| 1. | Tibbiy buyumning nomi/Name of a medical device
(Наименование изделия медицинского назначения) | |
| Davlat va ingliz tillarida/ In a state and English languages
(на государственном и английском языках) | ||
| 2. | Tibbiy buyumning modeli/ Model of a medical device
(Модель изделия медицинского назначения) | |
| Modeli/model
(модель) | ||
| Modifikatsiya/Modification
(модификация) | ||
| Butlovchi qismlar/ Accessories (комплектующие части) | ||
| Нajmi/ Packing
(объем) | ||
| Qo‘llash usuli/ application mode
(способприменения) | ||
| O‘ramdagi miqdori/ quantity in one pack (количествовупаковке) | ||
| 3. | O‘rami / Packing
(Упаковка) | |
| Birlamchi/ inner
(первичная) | ||
| Ikkilamchi/ outer
(вторичная) | ||
| 4. | Ariza beruvchi tashkilot nomi va davlati/ Name and country of applicant
(Наименование и страна заявителя) | |
| Davlat va ingliz tillarida / In a state and English languages
(на государственном и английском языках) | ||
| Pochta adresi/ Mail address
(Почтовыйадрес) | ||
| Tel., faks, e-mail/ Tel., Fax, E-mail
(тел., факс, e-mail) | ||
| Rahbar F.I.O./Full name of the Нead
(Ф.И.О. руководителя) | ||
| Ishlab chiqaruvchi tashkilot to‘g‘risida ma’lumot/ Data on firm-manufacturer
(Сведения об организации-производителе) | ||
| Davlat va ingliz tillarida /In a state and English languages
(на государственном и английском языках) | ||
| Yuridik manzili/ Legal address
(юридическийадрес) | ||
| Tel., faks, e-mail/ Tel., Fax, E-mail
(тел., факс, e-mail) | ||
| 6. | Tibbiy buyum ishlab chiqarish («in bulk» mahsulotni ishlab chiqarish, qadoqlash (birlamchi, ikkilamchi o‘rami), sifat nazoratini ko‘rsatgan holda) (kerak emasini chizib tashlang)/Data on manufacture of a medical device(manufacture of «in bulk» products, pre-packing, packing (inner, outer packs), specifying the quality control) (delete the unnecessary)
(Производство изделия медицинского назначения (производство «in bulk» продукции, фасовка, упаковка (первичная, вторичная упаковки), с указанием контроля качества) (вычеркнуть ненужное)) | |
| «in bulk» mahsulot/ «in bulk» production («in bulk» продукция) | ||
| Qadoqlash va (yoki) o‘rami / Pre-packing and (or) packing
(фасовка и (или) упаковка) | ||
| Tayyor mahsulot ishlab chiqaruvchisi/ Manufacturing of finished product (Производительготовогопродукта) | ||
| Tibbiy buyumni ishlab chiqaruvchisi va boshqa ishtirokchilari/ Manufacturer and other participants in manufacturing of a medical product
(Производитель и другие участники производства изделия медицинского назначения) | ||
| 7. | Ishlab chiqaruvchi davlatida va boshqa davlatlarda ro‘yxatdan o‘tkazilganligi (davlatlar ro'yxati)/Registration in the country of origin and other countries (list of countries)
(Регистрация в стране производства и других странах (список стран)) | |
| 8. | Tibbiy buyumlarning yaroqlilik muddati/ Shelf life of a medical product
(Срок годности изделия медицинского назначения) | |
| Yaroqlilik muddati/ Shelf life
(срок годности) | ||
| 9. | Tashish usuli/ Transport conditions
(Условиятранспортировки) | |
| 10. | Saqlash sharoiti/ Storage conditions
(Условияхранения) | |
| 11. | Tibbiy buyumlarni qo‘llash sohasi/Scope of application of a medical device
(Область применения изделия медицинского назначения) | |
To‘ldirilgan sana:
20 yil «_____»_________
Yuridik shaxs rahbarining F.I.O.
Imzo
Tashkilot muhri
Date of filling: (Дата заполнения):
«_____»_________ 20 г
Full name of the Нead
Signature
Organization stamp
(Ф.И.О. руководителя юридического лица Подпись Печать организации)
ПРИЛОЖЕНИЕ 3
к Положению о порядке регистрации лекарственных средств и изделий медицинского назначения и выдачи регистрационного удостоверения
Документы, представляемые для регистрации лекарственных средств*
| Лекарственное средство | ||||||
| лекарственный препарат | субстанция | лекарственное растительное сырье | гомеопатический препарат | МИБП | ||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| I. Общая документация | ||||||
| I | Содержание | |||||
| I A | Административные данные | |||||
| I A 1. | Заявление по утвержденной форме | + | + | + | + | + |
| I A 2. | Сертификат на фармацевтический продукт (СРР), выданный согласно рекомендации ВОЗ (заверенные нотариально)
при отсутствии: регистрационное удостоверение в стране производителя | + | - | + | + | + |
| I A 3. | Сертификат GMP (с указанием даты и результатов последней инспекции) | + | + | + | + | + |
| I A 4. | Копия лицензии на право осуществления фармацевтической деятельности | + | + | + | + | + |
| I A 5. | Если в производственном процессе участвуют несколько производителей, документы пунктов IА2, IА3, IА4 представляются на всех участников производства | + | + | + | + | + |
| I A 6. | Лицензионный договор (соглашение) на право производства (до истечения срока действия патента на оригинальный препарат) | + | - | - | + | + |
| I A 7. | Копия охранного документа (патента) (при наличии) | + | + | + | + | + |
| I A 8. | Документ, подтверждающий качество активного вещества (сертификат анализа субстанции от производителя, сертификат соответствия, протокол анализа, аналитический паспорт, входной контроль и др.) | + | + | + | + | + |
| I A 9. | Документ, подтверждающий качество готового продукта трех промышленных серий (сертификат анализа, протокол анализа и др.) | + | + | + | + | + |
| I A 10. | Документ о прионовой безопасности на вещества животного происхождения от производителя, справка об отсутствии губчатой энцефалопатии крупного рогатого скота (BSE/TSE) | + | + | - | + | + |
| I A 11. | Сведения об отказе в регистрации, отзыве с рынка компетентным органом или заявителем, о прекращении действия регистрационного удостоверения или приостановлении его компетентным органом (с указанием причины в случае наличия прецедентов) | + | + | + | + | + |
| I B | Краткая характеристика лекарственного средства (SmPC), маркировка и инструкция по применению | |||||
| I B 1. | **Краткая характеристика лекарственного средства (SmPC) | + | - | + | + | + |
| I B 2. | Копия инструкции по применению лекарственного средства, утвержденной в стране производителя, проект инструкции по применению на государственном и русском языках | + | * | + | + | + |
| I B 3. | Цветные макеты упаковок и маркировки на бумажном и электронном носителях | + | + | + | + | + |
| I C | Детальное описание фармакологического надзора и системы управления рисками при медицинском применении лекарственного препарата, предлагаемое заявителем | + | - | - | + | + |
| I D | Документ, подтверждающий наличие квалифицированного лица, ответственного за фармакологический надзор, для сбора и регистрации побочных реакций, выявляемых на территории Республики Узбекистан | + | - | + | + | + |
| I Е | Отчеты независимых экспертов по химической, фармацевтической и биологической документации, фармако-токсикологической документации, клинической документации (информация об основных свойствах лекарственного средства) | + | - | + | + | + |
| II. Химическая, фармацевтическая и биологическая документация | ||||||
| II | Содержание | + | + | + | + | + |
| II A | Состав | |||||
| II A 1. | Состав лекарственного средства (с указанием количества и квалификационной характеристики каждого ингредиента, в том числе состава покрытия и корпуса капсул) | + | - | + | + | + |
| II A 2. | Упаковка (краткое описание) | + | + | + | + | + |
| II A 3. | Фармацевтическая разработка (обоснование состава, выбора первичных упаковок и др.) | + | - | + | + | + |
| II B | Метод изготовления (схема технологического процесса, проект технологического регламента) | |||||
| II B 1. | Производственная формула | + | + | + | + | + |
| II B 2. | Производственный процесс | + | + | + | + | + |
| II B 3. | Контроль в процессе производства (операционный контроль) | + | + | + | + | + |
| II B 4. | Валидация процесса | + | + | + | + | + |
| II C | Методы контроля исходных материалов* | + | + | + | + | + |
| II C 1. | Активная субстанция* | |||||
| II C 1.1. | Нормативные документы (спецификации и методы контроля качества)* активных веществ | + | + | + | + | + |
| II C 1.2. | Научные данные* | + | + | + | + | + |
| II C 1.2.1. | Номенклатура* | + | + | + | + | + |
| II C 1.2.2. | Описание* | + | + | + | + | + |
| II C 1.2.3. | Производство* | + | + | + | + | + |
| II C 1.2.4. | Контроль качества в процессе производства (промежуточный контроль) | + | + | + | + | + |
| II C 1.2.5. | Химическая разработка | + | + | + | + | + |
| II C 1.2.6. | Примеси* | + | + | + | + | + |
| II C 1.2.7. | Анализ серии* | + | + | + | + | + |
| II C 1.2.8. | Сведения по изучению стабильности, отчеты и срок годности | + | + | + | + | + |
| II C 2. | Вспомогательные вещества | |||||
| II C 2.1. | Нормативные документы (спецификации и установленные методы контроля качества) вспомогательных веществ | + | - | - | + | + |
| II С 2.2. | Научные данные | + | - | - | + | + |
| II С 2.3. | Анализ серии* | |||||
| II C 3. | Упаковочный материал (внутренняя/внешняя упаковка) | + | + | + | + | + |
| II C 3.1. | Спецификации и установленные методы контроля качества упаковочных материалов и сертификаты качества | + | + | + | + | + |
| II C 3.2. | Научные данные | + | + | + | + | + |
| II D | Методы контроля качества промежуточных продуктов | + | + | + | + | + |
| II E | Методы контроля качества конечного продукта | |||||
| II E 1. | Спецификации и методы контроля качества с аутентичным переводом на русский язык | + | + | + | + | + |
| II E 1.1. | Нормативный документ лекарственного средства | + | + | + | + | + |
| II E 1.2. | Сведения о стандартах | + | + | + | + | + |
| II E 1.3. | Пояснительная записка к нормативному документу | + | + | + | + | + |
| II E 1.4. | При продлении срока регистрации копия утвержденного нормативного документа | + | + | + | + | + |
| II E 2. | Научные данные | + | + | + | + | + |
| II E 2.1. | Валидация аналитических методов и комментарии, стандарты (рабочие стандарты) | + | + | + | + | + |
| II E 2.2. | Анализ серии | + | + | + | + | + |
| II F | Стабильность | |||||
| II F 1. | Методы изучения стабильности | + | + | + | + | + |
| II F 2. | Результаты испытания стабильности не менее чем на 3-х промышленных или опытно-промышленных (пилотных) сериях (данные и отчеты стресс-тестов и долгосрочных испытаний в реальном времени) | + | + | + | + | + |
| II G | Сведения о профиле растворения (для твердых дозированных лекарственных форм) | + | - | - | + | + |
| II Н | Данные по биодоступности, биоэквивалентности (для генериков), для парентеральных форм генериков — данные по безопасности и эффективности | + | - | - | + | + |
| II K | Данные контроля на животных | - | - | - | - | - |
| II L | Данные по вероятной опасности для окружающей среды для препаратов, содержащих генетически измененные организмы | + | + | - | - | + |
| II M | Периодический обновляемый отчет по безопасности | + | - | + | + | + |
| II Q | Дополнительная информация, подтверждающая качество | + | + | + | + | + |
| III. Фармакологическая и токсикологическая документация | ||||||
| III | Содержание | + | + | + | + | + |
| III A | Токсичность при однократном введении и введении повторных доз (острая, подострая, хроническая токсичность) | + | - | - | + | + |
| III B | Влияние на репродуктивную функцию
Данные по тератогенности, гонадотоксичности и эмбриотоксичности | + | - | - | - | + |
| III C | *Данные по мутагенности | + | - | - | - | + |
| III D | *Данные по канцерогенности | + | - | - | - | + |
| III Е | Фармакодинамика (специфическое действие, общие фармакологические свойства (для медицинских иммунобиологических препаратов — реактогенность, специфическая активность) | + | - | - | + | + |
| III F | Фармакокинетика (всасывание, распределение, метаболизм, выделение) | + | - | - | - | + |
| III G | Данные по биоэквивалентности (для генериков), для МИБП — сравнительная характеристика | + | - | - | - | + |
| III Н | *Данные о местно-раздражающем действии (для МИБП, в том числе вакцин — иммуногенность) | + | - | - | - | + |
| III K | *привыкание к лекарственному средству и кумулятивные свойства | + | - | + | - | + |
| III L | *Антигенность | + | - | - | - | + |
| III М | Дополнительная информация, подтверждающая безопасность | + | + | + | + | + |
| IV. ** Клиническая документация | ||||||
| IV | Содержание | + | + | + | + | + |
| IV A | Данные по клинической фармакологии (фармакодинамика, фармакокинетика) (in vivo для медицинских иммунобиологических препаратов — данные по эпидемиологической, клинической, иммунологической эффективности) | + | - | - | + | + |
| IV B | Результаты клинических испытаний, научные публикации, отчеты | + | - | - | + | + |
| IV С | Данные пострегистрационного опыта (если таковой имеется) | + | - | - | + | + |
| IV D | Дополнительная информация, подтверждающая эффективность и безопасность, качество, клиническая эффективность и безопасность и другая дополнительная информация после регистрации | + | - | - | + | + |
Примечания:
*Документы, указанные в перечне, требуются с учетом происхождения, свойств, особенностей способа получения/производства лекарственных средств; если отдельные части документации не включены в регистрационное досье, то следует в соответствующем месте указать причину под соответствующим заголовком.
**При наличии.
***Для лекарственных препаратов животного происхождения в разделе IIС1 должна быть представлена следующая дополнительная информация:
- данные относительно вида, возраста, рациона и географического региона происхождения животных, от которых получено сырье;
- данные о характере (категории) ткани, из которой получается сырье для производства лекарственного средства, с точки зрения ее опасности относительно содержания прионов;
- технологическая схема обработки сырья с указанием экстрагентов, температурного режима и т. п.;
- методы контроля исходного сырья.
ПРИЛОЖЕНИЕ 3а
к Положению о порядке регистрации лекарственных средств и изделий медицинского назначения и выдачи регистрационного удостоверения
ПЕРЕЧЕНЬ
документов, представляемых для регистрации изделия медицинского назначения
- Список представляемых документов.
- Доверенность от предприятия — производителя на должностное лицо, уполномоченное для ведения делопроизводства по регистрации.
- Общая информация об изделии медицинского назначения.
- Регистрационное удостоверение изделия медицинского назначения (если изделие медицинского назначения было ранее зарегистрировано).
- Нормативный документ, включающий в себя порядок и методы испытания изделия медицинского назначения, стандарт на аналогичную продукцию.
- Для продукции серийного производства проекты стандарта организации/фармакопейной статьи, технологического регламента, технологической инструкции.
- Паспорт на медицинскую технику, аналитический паспорт на изделие медицинского назначения.
- Техническое описание медицинской техники.
- Информация о лабораторных испытаниях, технических испытаниях, доклинических исследованиях и клинических (медицинских) испытаниях.
- Методы испытаний, программы и протоколы (для стерильных изделий медицинского назначения — испытания, проведенные за последний год) на изделия медицинского назначения.
- Информация о соответствии изделия медицинского назначения международным стандартам (при наличии).
- Методы поверки медицинской техники (для измерительных средств).
- Информация об отсутствии инфекционных агентов в «in vitro» — диагностических средствах, приготовленных из крови человека.
- Информация о стабильности при хранении и транспортировке «in vitro» — диагностических средств.
- Иллюстрированные рекламные материалы, проспекты, каталоги, фото размером не менее 13х18см.
- Образец изделия медицинского назначения (в достаточном количестве для проведения испытаний согласно нормативному документу).
- Протокол анализов представленной серии изделия медицинского назначения.
- Цветные графические макеты первичной и вторичной упаковки.
Выше указанные документы представляются в Главное управление в двух экземплярах.
Копии документов должны быть читабельными и заверены печатью и подписью руководителя организации.
ПРИЛОЖЕНИЕ 4
к Положению о порядке регистрации лекарственных средств и изделий медицинского назначения и выдачи регистрационного удостоверения






Номер документа: № 352
Дата принятия: 22.12.2014